
L’Associazione conSLAncio Onlus di Terracina (LT), fondata da Andrea Zicchieri, Membro dell’International Alliance of ALS/MND Associations (Alleanza Internazionale delle Associazioni SLA/MND), era presente al 33° Simposio Internazionale sulla SLA/MND Virtuale, 6-9 Dicembre 2022. Il Simposio è la più grande conferenza al mondo sulla Sclerosi Laterale Amiotrofica (SLA) organizzata dalla Motor Neurone Disease Association. L’edizione 2022 ha ospitato oltre 1.300 delegati da 45 nazioni, incluso medici, ricercatori, pazienti, caregiver ed organizzazioni della comunità SLA internazionale.
conSLAncio vi presenta il più comprensivo resoconto del simposio in Italiano.
.
 Durante l’inaugurazione dell’evento, l’International Alliance of ALS/MND Associations ha conferito il Premio Forbes Norris Award 2022 al nostro consulente scientifico prof. Richard Bedlack, Direttore della Clinica SLA presso la Duke University Medical Center, Durham, North Carolina, Stati Uniti. Questo premio rappresenta il massimo riconoscimento globale nel campo clinico sulla SLA e premia la combinazione di due maggiori qualità: la cura e l’assistenza dei pazienti ed il progresso delle conoscenze scientifiche. Il prof. Bedlack è stato premiato per l’attività clinica e di ricerca riconosciute a livello internazionale per diverse ragioni fondamentali che ne giustificano l’impatto. Bedlack è convinto che tutti coloro che convivono con la SLA/MND debbano ricevere un’assistenza della massima qualità possibile. È con questa filosofia in mente che ha creato e dirige la Duke ALS Clinic. Aderendo alle migliori pratiche, offre alle famiglie le risorse di un’ampia équipe interdisciplinare, presentando tutte le opzioni terapeutiche disponibili, in modo che possano affrontare la malattia nel modo migliore per loro. Guidare e partecipare a iniziative di ricerca che creano una migliore comprensione e migliori trattamenti per la SLA/MND è parte integrante dell’approccio del prof. Bedlack alla cura dei pazienti.
Durante l’inaugurazione dell’evento, l’International Alliance of ALS/MND Associations ha conferito il Premio Forbes Norris Award 2022 al nostro consulente scientifico prof. Richard Bedlack, Direttore della Clinica SLA presso la Duke University Medical Center, Durham, North Carolina, Stati Uniti. Questo premio rappresenta il massimo riconoscimento globale nel campo clinico sulla SLA e premia la combinazione di due maggiori qualità: la cura e l’assistenza dei pazienti ed il progresso delle conoscenze scientifiche. Il prof. Bedlack è stato premiato per l’attività clinica e di ricerca riconosciute a livello internazionale per diverse ragioni fondamentali che ne giustificano l’impatto. Bedlack è convinto che tutti coloro che convivono con la SLA/MND debbano ricevere un’assistenza della massima qualità possibile. È con questa filosofia in mente che ha creato e dirige la Duke ALS Clinic. Aderendo alle migliori pratiche, offre alle famiglie le risorse di un’ampia équipe interdisciplinare, presentando tutte le opzioni terapeutiche disponibili, in modo che possano affrontare la malattia nel modo migliore per loro. Guidare e partecipare a iniziative di ricerca che creano una migliore comprensione e migliori trattamenti per la SLA/MND è parte integrante dell’approccio del prof. Bedlack alla cura dei pazienti.
Il prof. Bedlack si è affermato come leader del settore grazie al suo coinvolgimento in sperimentazioni multicentriche. Dai modelli di studio innovativi alla formazione di ambasciatori della ricerca attraverso il NEALS Clinical Research Learning Institute, Bedlack è stato in prima linea nelle iniziative di sostegno e formazione per ampliare le opportunità di ricerca e aumentare la partecipazione agli studi, con l’obiettivo di creare un mondo libero dalla SLA/MND. Durante questo periodo, il prof. Bedlack ha istituito una clinica per la SLA di livello mondiale, ha contribuito in modo significativo alla ricerca clinica e si è impegnato in iniziative che hanno portato a cambiamenti positivi in termini di cure, istruzione, benefici e ricerca. Il prof. Bedlack incarna veramente il termine “cura del paziente” attraverso l’empatia, la compassione e l’impegno che dimostra con ogni paziente che visita. La sua incrollabile dedizione alla causa della SLA/MND è evidente in ogni aspetto del suo lavoro e lo rende un’ispirazione per tutti coloro che sono affetti da questa malattia in tutto il mondo.
Uno dei suoi contributi pubblici più importanti è la creazione del programma ALSUntangled. Si tratta di un’enorme risorsa per la comunità SLA, che fornisce revisioni scientifiche di trattamenti alternativi ed off-label per aiutare le persone a prendere decisioni informate sulle loro cure. È possibile ascoltare le puntate audio di ALSUntangled in Italiano sul sito di conSLAncio, disponibile dal 2019, cliccando qui.
<<>>
 La Dott.ssa Chen Eitan, PhD, Borsista post-dottorato del Dipartimento di Genetica Molecolare e Neuroscienze Molecolare presso il Weizmann Institute of Science, Tel-Aviv, Israele ha vinto il 14° Premio Paulo Gontijo Award 2022. Il Premio Paulo Gontijo, noto anche come PG Award, premia dal 2007 giovani ricercatori di tutto il mondo impegnati nella ricerca delle cause e delle cure della SLA. Si tratta di un premio in onore di Paulo Gontijo, forte sostenitore della ricerca cui è dedicato anche l’Istituto Paulo Gontijo.
La Dott.ssa Chen Eitan, PhD, Borsista post-dottorato del Dipartimento di Genetica Molecolare e Neuroscienze Molecolare presso il Weizmann Institute of Science, Tel-Aviv, Israele ha vinto il 14° Premio Paulo Gontijo Award 2022. Il Premio Paulo Gontijo, noto anche come PG Award, premia dal 2007 giovani ricercatori di tutto il mondo impegnati nella ricerca delle cause e delle cure della SLA. Si tratta di un premio in onore di Paulo Gontijo, forte sostenitore della ricerca cui è dedicato anche l’Istituto Paulo Gontijo.
Il manoscritto della Dott.ssa Eiten intitolato, “Whole-genome sequencing reveals that variants in the Interleukin 18 Receptor Accessory Protein 3’ UTR protect against ALS” (“Il sequenziamento genomico rivela che varianti genetiche al 3’ UTR della proteina accessoria del recettore per l’interleuchina 18 protegge control la SLA”) si è distinto vincendo il Premio Gontijo. È stato pubblicato sulla rivisita Nature Neuroscience 25, 433–445 (2022), https://doi.org/10.1038/s41593-022-01040-6. La Dott.ssa Eiten ha presentato il suo lavoro alla comunità scientifica presente; come riconoscimento ha ricevuto una medaglia d’oro e un finanziamento scientifico di 10.000 dollari.
<<>>
La Dott.ssa Sally Light, Presidente/CEO della Motor Neurone Disease Association ha vinto il Premio Humanitarian Award 2022 dall’International Alliance of ALS/MND Associations. Questo prestigioso riconoscimento onora la dedizione della Dott.ssa Light nel migliorare la qualità della vita delle persone affette da SLA/MND, in modo non-scientifico, in tutto il mondo. La Dott.ssa Light è andata in pensione dopo dieci anni di servizio nel 2022.
<<>>
Il 33° Simposio Internazionale sulla SLA/MND celebra il terzo anno consecutivo in cui la nostra associazione ha presentato un poster al congresso. Abbiamo presentato la base del nostro nuovo seminario virtuale in arrivo su conSLAncio.
Il poster presentato da conSLAncio
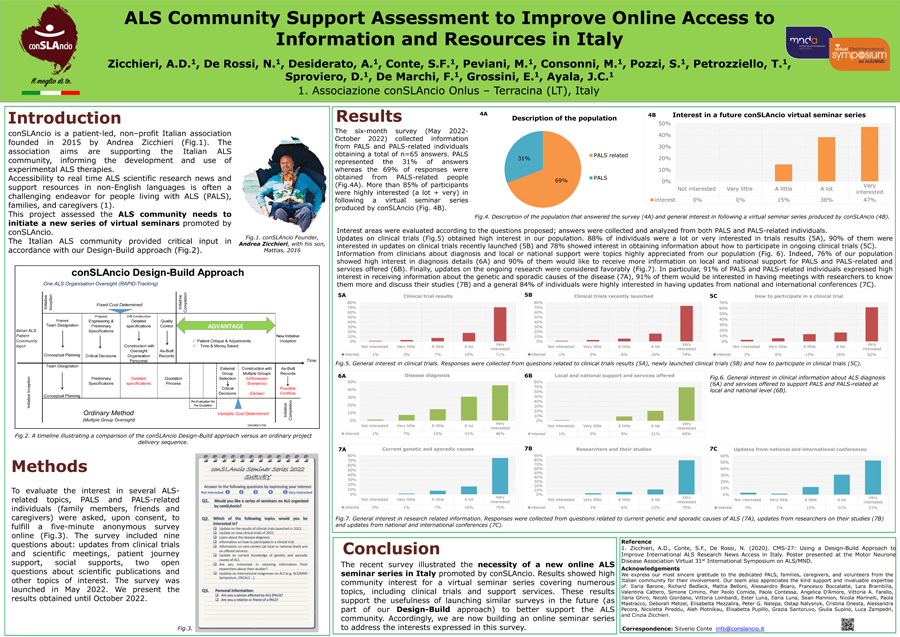 CMS-22: Zicchieri A.D., De Rossi N., Desiderato A., Conte S.F., Peviani, M., Consonni, M., Pozzi, S., Petrozziello, T., Sproviero, D., De Marchi F., Grossini, E., Ayala J.C., “ALS Community Support Assessment to Improve Online Access to Information and Resources in Italy” (“Valutazione dei bisogni di supporto nella comunità SLA per migliorare l’accesso online alle informazioni e risorse in Italia”).
CMS-22: Zicchieri A.D., De Rossi N., Desiderato A., Conte S.F., Peviani, M., Consonni, M., Pozzi, S., Petrozziello, T., Sproviero, D., De Marchi F., Grossini, E., Ayala J.C., “ALS Community Support Assessment to Improve Online Access to Information and Resources in Italy” (“Valutazione dei bisogni di supporto nella comunità SLA per migliorare l’accesso online alle informazioni e risorse in Italia”).
L’abstract:
Premessa: L’accessibilità ai progressi della ricerca scientifica e alle risorse di supporto riguardanti la SLA spesso rappresentano una sfida per le persone affette da questa malattia (PALS), le loro famiglie e i loro assistenti (1), ostacolati anche dalla diffusione in lingua inglese dei comunicati della comunità scientifica.
Scopo: Lo studio in oggetto si è posto come obiettivo valutare i bisogni dei PALS, delle loro famiglie e dei loro assistiti per lanciare una nuova serie di seminari virtuali che possano rispondere ad esigenze specifiche. Questa iniziativa è stata promossa da conSLAncio Onlus, un’associazione italiana sulla SLA guidata da un paziente che opera con un team multidisciplinare di esperti clinici e ricercatori.
Metodi: Ai PALS e ai familiari è stato chiesto di compilare un questionario online, anonimo, della durata di cinque minuti, per avere un riscontro del livello di interesse verso alcuni argomenti legati alla SLA che sarebbero poi stati affrontati in un nuovo ciclo di seminari virtuali. Il questionario comprendeva nove domande inerenti aggiornamenti sugli studi clinici e riunioni scientifiche, esperienze del paziente e altri supporti sociali, alle quali era richiesto di rispondere attribuendo un valore corrispondente al livello di interesse (da 1 = non interessato, a 5 = molto interessato). Infine, il questionario terminava lasciando due domande aperte con possibilità di chiedere chiarimenti in merito alle pubblicazioni scientifiche e ad altri temi di interesse non citati precedentemente. Il sondaggio è stato lanciato a maggio 2022.
Risultati: Ad ottobre 2022, 65 partecipanti hanno risposto al sondaggio: Il 31% erano PALS e il 69% era rappresentato da persone (familiari, amici) legati ai PALS. Complessivamente, l’85% dei partecipanti ha espresso grande interesse per la proposta di una nuova serie di seminari virtuali. La maggior parte dei partecipanti (90%) ha espresso interesse per gli aggiornamenti dei nuovi studi clinici e per ricevere maggiori informazioni sul supporto locale e nazionale per i PALS e sui servizi offerti. Anche aggiornamenti sulla ricerca in corso sono stati considerati favorevolmente.
Discussione: Nel complesso, grazie a questo sondaggio, abbiamo potuto riscontrare un grande interesse da parte della comunità italiana per la proposta di sviluppare una nuova serie di seminari virtuali sulla SLA. La maggior parte dei partecipanti vorrebbe ricevere informazioni su studi clinici, approcci terapeutici e attività dai ricercatori, suggerendo la necessità di una discussione con esperti. Di conseguenza, stiamo costruendo una serie di seminari online per affrontare gli interessi espressi in questo sondaggio utilizzando un approccio di tipo design-build (progettazione-costruzione) promossa da conSLAncio (1). Scarica il poster di conSLAncio in fondo della pagina sotto ‘Risorse Correlate’.
Bibliografia: (1) Zicchieri, A.D., Conte, S.F., De Rossi, N. (2020). CMS-27: Using a Design-Build Approach to Improve International ALS Research News Access in Italy. Poster presented at the Motor Neurone Disease Association Virtual 31st International Symposium on ALS/MND.
conSLAncio desidera rivolgere una standing ovation alla straordinaria dedizione di pazienti, famiglie, assistenti, volontari della comunità italiana e di tutto il mondo che ci aiutano nella nostra missione:
Ilaria Barone, Richard Bedlack, Mattia Belloni, Alessandro Boaro, Francesco Boccalatte, Lara Brambilla, Valentina Cattero, Simone Cimino, Pier Paolo Comida, Paola Contessa, Angelica D’Amore, Vittoria A. Farallo, Ilaria Ghiro, Nicolò Giordano, Vittoria Lombardi, Ester Luna, Ilaria Luna, Sean Mannion, Nicola Marinelli, Paola Mastracco, Deborah Metzel, Elisabetta Mezzalira, Peter G. Nalepa, Ostap Nalysnyk, Cristina Onesta, Alessandra Pecora, Nicoletta Pireddu, Aleh Plotnikau, Elisabetta Pupillo, Grazia Santoruvo, Giulia Supino, Luca Zampedri e Cinzia Zicchieri.
<<>>
C’erano 25 presentazioni su piattaforma e oltre 250 poster, presentando le ultime novità scientifiche della SLA/MND. Segnaliamo alcuni studi clinici e poster che hanno fornito un aggiornamento importante.
 C2: La nostra consulente scientifica Dott.ssa Elisabetta Pupillo, PharmD, PhD dell’Istituto “Mario Negri” di Milano ha presentato il suo studio nella sessione plenaria del Simposio: “Risultati di uno studio randomizzato, in doppio cieco, controllato con placebo sull’uso di RNS60 nelle persone con SLA”.
C2: La nostra consulente scientifica Dott.ssa Elisabetta Pupillo, PharmD, PhD dell’Istituto “Mario Negri” di Milano ha presentato il suo studio nella sessione plenaria del Simposio: “Risultati di uno studio randomizzato, in doppio cieco, controllato con placebo sull’uso di RNS60 nelle persone con SLA”.
La SLA è una malattia neurodegenerativa fatale per la quale sono disponibili limitati trattamenti. RNS60 è un prodotto sperimentale immunomodulatore e neuroprotettivo che è risultato efficace in modelli animali di SLA e di altre malattie neurodegenerative. In studi precedenti, la somministrazione è risultata sicura e ben tollerata nei pazienti SLA.
La Dott.ssa Pupillo ed il suo gruppo hanno condotto uno studio di Fase II, multicentrico, randomizzato, in doppio cieco, controllato con placebo, a gruppi paralleli. I partecipanti sono stati randomizzati a ricevere RNS60 o placebo per 24 settimane. I pazienti sono stati seguiti per ulteriori 24 settimane dopo il termine del trattamento. L’obiettivo principale era quello di misurare gli effetti di RNS60 su biomarcatori selezionati di infiammazione e neurodegenerazione nel sangue periferico, mentre gli obiettivi secondari comprendevano la valutazione dell’effetto di RNS60 su peggioramento clinico (scala funzionale ALSFRS – revisionata (ALSFRS-R) e autosufficienza), funzione respiratoria (capacità vitale forzata (FVC)), qualità della vita (ALSAQ-40), sopravvivenza, tollerabilità e sicurezza.
Sono stati inclusi 147 soggetti, di cui 74 hanno ricevuto RNS60 e 73 hanno ricevuto il placebo. I risultati non hanno rivelato alcuna differenza nei livelli dei biomarcatori misurati. Tuttavia, è stato osservato un rallentamento nel tasso medio di declino della FVC nel gruppo RNS60, con una differenza di 0.41 punti a settimana. Anche nel dominio della scala sulla qualità di vita ALSAQ-40 relativo al mangiare e bere si è osservato un peggioramento rallentato nel gruppo RNS60, con una differenza di 0.19 punti a settimana. Gli effetti collaterali osservati erano simili nei due gruppi. Tuttavia, ulteriori studi sono necessari per validare gli effetti positivi di RNS60 sulla funzione respiratoria.
<<>>
 C3: Il prof. Gilbert Bensiman del Centre Hospitalier Universitaire (CHU) di Nimes, Francia ha presentato i dati di sicurezza e efficacia a basso dosaggio dell’interleuchina 2 (IL-2) nella SLA ottenuti dallo MIROCALS. Si ritiene che l’infiammazione e le alterazioni del sistema immunitario svolgano un ruolo nella progressione della SLA/MND. È stato dimostrato che la somministrazione di IL-2 a basse dosi aumenta il numero di cellule immunitarie che riducono l’infiammazione. Pertanto, è stato ipotizzato che basse dosi di IL-2 possano rallentare la progressione della SLA. Lo studio MIROCALS (Modifying Immune Response and OutComes in ALS), uno studio randomizzato di fase 2 controllato con placebo, è stato condotto su 220 partecipanti provenienti da 17 centri SLA/MND nel Regno Unito e in Francia. I partecipanti sono stati seguiti per una fase preliminare di 3 mesi, prima di essere randomizzati a ricevere IL-2 o placebo per 18 mesi. L’obiettivo primario dello studio consisteva nel valutare l’effetto dell’IL-2 sulla sopravvivenza. Obiettivi secondari includevano la valutatione di qualità della vita, funzionalità e biomarcatori di SLA/MND nel sangue e nel liquido cerebrospinale.
C3: Il prof. Gilbert Bensiman del Centre Hospitalier Universitaire (CHU) di Nimes, Francia ha presentato i dati di sicurezza e efficacia a basso dosaggio dell’interleuchina 2 (IL-2) nella SLA ottenuti dallo MIROCALS. Si ritiene che l’infiammazione e le alterazioni del sistema immunitario svolgano un ruolo nella progressione della SLA/MND. È stato dimostrato che la somministrazione di IL-2 a basse dosi aumenta il numero di cellule immunitarie che riducono l’infiammazione. Pertanto, è stato ipotizzato che basse dosi di IL-2 possano rallentare la progressione della SLA. Lo studio MIROCALS (Modifying Immune Response and OutComes in ALS), uno studio randomizzato di fase 2 controllato con placebo, è stato condotto su 220 partecipanti provenienti da 17 centri SLA/MND nel Regno Unito e in Francia. I partecipanti sono stati seguiti per una fase preliminare di 3 mesi, prima di essere randomizzati a ricevere IL-2 o placebo per 18 mesi. L’obiettivo primario dello studio consisteva nel valutare l’effetto dell’IL-2 sulla sopravvivenza. Obiettivi secondari includevano la valutatione di qualità della vita, funzionalità e biomarcatori di SLA/MND nel sangue e nel liquido cerebrospinale.
L’IL-2 è risultata sicura e ben tollerata, gli effetti collaterali sono stati minimi sia con il trattamento che con il placebo. Analizzando l’intera popolazione dello studio, il trattamento con IL-2 ha mostrato una modesta riduzione del rischio di morte, non statisticamente significativa, per i soggetti in trattamento nei 21 mesi dello studio. Tuttavia, quando i partecipanti sono stati esaminati in sottogruppi definiti in base a specifici biomarcatori, l’effetto sulla sopravvivenza è risultato statisticamente significativo nei partecipanti che hanno ricevuto IL-2 con una riduzione di oltre il 40% del rischio di morte a 21 mesi per l’80% della popolazione complessiva dello studio. Ulteriori studi sono attualmente in corso per capire come il trattamento con IL-2 influisca sulla progressione della malattia ed inoltre, i ricercatori che hanno condotto lo studio stanno discutendo con gli enti regolatori dei farmaci per determinare le fasi successive.
<<>>
 C4: Il prof. Steve Vucic dell’Università di Sydney, Australia ha presentato gli ultimi dati dello studio RESCUE-ALS sul trattamento CNM-Au8. Si ritiene che alterazioni nella produzione cellulare di energia nella SLA riduca la capacità dei neuroni di funzionare correttamente. CNM-Au8 è una sospensione orale contenente nanocristalli d’oro che contribuisce ad aumentare la produzione di energia, migliorando la sopravvivenza, la funzione e la comunicazione dei neuroni nelle persone che vivono con SLA/MND.
C4: Il prof. Steve Vucic dell’Università di Sydney, Australia ha presentato gli ultimi dati dello studio RESCUE-ALS sul trattamento CNM-Au8. Si ritiene che alterazioni nella produzione cellulare di energia nella SLA riduca la capacità dei neuroni di funzionare correttamente. CNM-Au8 è una sospensione orale contenente nanocristalli d’oro che contribuisce ad aumentare la produzione di energia, migliorando la sopravvivenza, la funzione e la comunicazione dei neuroni nelle persone che vivono con SLA/MND.
Uno studio di fase 2 su CNM-Au8 è stato condotto su 45 persone con SLA/MND e i partecipanti sono stati assegnati in modo casuale al gruppo placebo o CNM-Au8 per 36 settimane. L’obiettivo primario dello studio era la valutazione di un indice chiamato “Motor Unit Number Index” (MUNIX), una misura neurofisiologica quantitativa che può essere utilizzata per stimare il numero di connessioni tra motoneuroni e muscoli. Obiettivi secondari comprendevano, invece, la valutazione di funzione respiratoria tramite la capacità vitale forzata (FVC) e progressione della malattia tramite l’ALSFRS-R. Lo studio non ha raggiunto l’obiettivo primario, ma sono emerse tendenze positive che suggeriscono che il CNM-Au8 potrebbe essere utile per la SLA, in quanto sono state registrate riduzioni statisticamente significative della progressione della SLA, cosi come un potenziale beneficio in termini di sopravvivenza a lungo termine.
I partecipanti allo studio RESCUE-ALS hanno avuto la possibilità di partecipare anche all’estensione dello studio in aperto, nel quale tutti i partecipanti hanno potuto assumere CNM-Au8. Per verificare se l’assunzione di CNM-Au8 per un periodo prolungato avesse maggiori benefici, i partecipanti sono stati seguiti per ulteriori 48 settimane. I risultati di tale studio hanno rivelato che CNM-Au8 potrebbe migliorare la sopravvivenza se assunto per un lungo periodo: infatti, è stata osservata una riduzione del 70% del rischio di more per i partecipanti che hanno cominciato la terapia con CNM-Au8 durante RESCUE-ALS rispetto ai partecipanti che erano nel gruppo del placebo durante la prima fase dello studio.
Ulteriori studi sono necessari per validare l’utlizzo terapeutico di CNM-Au8 e, di fatti, si sta pianificando uno studio di fase 3. CNM-Au8 è anche del platform trial presso l’Healey Center per la SLA al Massachusetts General Hospital in Boston, USA. Sfortunatamente, sia l’obiettivo primario (valutazione della progressione della malattia sulla base di ALSFRS-R) che gli obiettivi secondari (valutazione della progressione della malattia sulla base di CAFS e SVC) non sono stati raggiunti in 24 settimane di trattamento. Il trattamento con CNM-AU8 è comunque risultato ben tollerato e sicuro.
<<>>
 C5: Il prof. Timothy Miller della Washington University ha presentato i risultati dello studio Fase 3 VALOR e studio di estensione in aperto sul Tofersen per la SLA-SOD1. Diverse mutazioni genetiche sono responsabili dello sviluppo della SLA, incluse mutazioni nel gene codificante per la superossido dismutasi 1 (SOD1). Tofersen è un oligonucleotide antisenso che è stato progettato per indirizzare le istruzioni genetiche per SOD1 mutato in modo da ridurne la quantità prodotta.
C5: Il prof. Timothy Miller della Washington University ha presentato i risultati dello studio Fase 3 VALOR e studio di estensione in aperto sul Tofersen per la SLA-SOD1. Diverse mutazioni genetiche sono responsabili dello sviluppo della SLA, incluse mutazioni nel gene codificante per la superossido dismutasi 1 (SOD1). Tofersen è un oligonucleotide antisenso che è stato progettato per indirizzare le istruzioni genetiche per SOD1 mutato in modo da ridurne la quantità prodotta.
Lo studio VALOR era uno studio randomizzato di fase 3 controllato con placebo che ha incluso 108 partecipanti. I partecipanti sono stati assegnati in modo casuale al gruppo di trattamento con Tofersen o al gruppo placebo per 28 settimane. L’esito primario dello studio consisteva nel misurare l’effetto del Tofersen sulla funzionalità mediante l’ALSFRS-R. I risultati dello studio VALOR non hanno evidenziato differenze significative nella funzionalità tra i pazienti trattati con Tofersen e quelli trattati con placebo. Tuttavia, sono emersi alcuni segnali promettenti: infatti, il trattamento con Tofersen ha ridotto i livelli di SOD1 e ha contribuito a diminuire i livelli di un biomarcatore chiamato catena leggera dei neurofilamenti, NfL (un marcatore del danno neuronale), suggerendo una diminuzione dell’attività e della progressione della malattia.
Al termine dello studio, i partecipanti sono stati invitati a prendere parte all’estensione in aperto e sono stati seguiti per ulteriori 52 settimane in modo da verificare gli effetti di un trattamento prolungato. I risultati hanno dimostrato che Tofersen potrebbe essere efficace se assunto per un periodo di tempo più lungo. Infatti, l’assunzione del Tofersen per un periodo più lungo ha portato a miglioramenti o stabilizzazioni sia della forza muscolare che della funzione respiratoria. È stato inoltre rilevato che Tofersen potrebbe avere un effetto positivo sul peso corporeo: i soggetti che hanno assunto Tofersen più a lungo sono infatti aumentati di peso o non hanno perso ulteriore peso. Ciò suggerisce che Tofersen potrebbe essere utile per le persone affette da SLA-SOD1 e, pertanto, è stata presentata la domanda di registrazione di un nuovo farmaco per Tofersen alla FDA statunitense e la decisione è prevista per il 25 aprile 2023.
AMX0035 (CENTAUR e PHOENIX)
AMX0035 è una terapia combinata di fenilbutirrato di sodio (PB) e taurusodiolo (TURSO o TUDCA) recentemente approvata per il trattamento della SLA negli Stati Uniti e in Canada. I due composti mirano a colpire la malattia in modo leggermente diverso: il PB riduce lo stress del reticolo endoplasmatico, mentre il TUDCA blocca le principali vie di morte cellulare. La combinazione dei due farmaci riduce la morte delle cellule nervose nelle persone che vivono con la SLA.
AMX0035 è stato testato in uno studio clinico di Fase 2 chiamato CENTAUR. Questo studio ha rilevato che AMX0035 rallentava modestamente i sintomi della malattia e ha evidenziato una differenza di 6,5 mesi nella sopravvivenza di coloro che hanno ricevuto AMX0035 rispetto a coloro che hanno ricevuto il placebo. È attualmente in corso uno studio di Fase 3 denominato PHOENIX che include una popolazione più eterogenea di pazienti SLA seguiti per un periodo di tempo più lungo rispetto allo studio CENTAUR. Le modalità dello studio PHOENIX sono state descritte nel poster CLT-24. Lo studio continuerà le valutazioni di sicurezza ed efficacia di PB and TURSO nella SLA sia in Europa che negli Stati Uniti.
AMX0035 è stato discusso anche nel poster CLT-01, nel quale un gruppo di revisori indipendenti ha valutato effetti cardiaci collaterali dovuti all’assunzione di AMX0035 escludendo ogni possibile effetto avverso a livello cardiaco dovuto a PB o TURSO.
Il poster CLT-07 parla anche del AMX0035, fornendo aggiornamenti e insegnamenti dal programma di accesso allargato. I programmi di accesso allargato consentono l’accesso a terapie sperimentali a persone che non erano eleggibili per la sperimentazione clinica.
PHOENIX (NCT05021536) ha 8 centri in Italia; per maggiori informazioni su questo studio, clicca qui.
WVE-004 (FOCUS-C9) WVE-004 è un oligonucleotide antisenso (ASO) progettato per ridurre il numero di ripetizioni nucleotidiche nella sequenza genetica codificante per C9orf72, il gene più comunemente mutato nella SLA. Risultato di queste ripetizioni nucleotiche è l’accumulo di proteine dipeptidiche come Poly(GP).
Lo studio clinico di fase 1/2 FOCUS-C9 mira a valutare la sicurezza, la tollerabilità e il comportamento di WVE-004 all’interno dell’organismo (effetti farmacocinetici e farmacodinamici) sia nei pazienti SLA che nei pazienti affetti da demenza frontotemporale (FTD). Wave Life Sciences ha annunciato i risultati intermedi dello studio nel corso del 2022. Questi risultati hanno dimostrato una riduzione dei livelli di Poly(GP) con basse e singole dosi di WVE-004, indicando che il farmaco riconosce l’esatto bersaglio molecolare. Questi risultati sono stati presentati nel poster CLT-04.
FOCUS-C9 sta ancora reclutando nel Regno Unito, Irlanda, Olanda, Belgio e Svezia in Europa.
Tegoprubart (AT-1501)
Tegoprubart è un anticorpo terapeutico che riconosce una proteina chiamata ligando del CD40 (CD40L). Il CD40L è implicato nella regolazione della risposta immunitaria, che può causare infiammazione del midollo spinale. È stato riportato che la via di trasduzione del segnale del CD40L è iperfunzionante nelle persone affette da SLA. Pertanto, è stato ipotizzato che l’uso del Tegoprubart per inibire CD40L possa bloccare o ritardare l’attivazione della risposta immunitaria infiammatoria.
Tegoprubart è in fase di sperimentazione in uno studio clinico di fase 2° atto a validarne sicurezza e tollerabilità a 4 diverse dosi. La sperimentazione di diverse dosi in uno studio clinico è una fase fondamentale della sperimentazione di un nuovo potenziale farmaco per determinare il dosaggio corretto. Lo studio ha dimostrato che Tegoprubart è sicuro e ben tollerato ed ha un effetto dose-dipendente. I risultati dello studio sono stati descritti nel poster CLT-02.
QRL-201
QRL-201-01 è uno studio clinico multi-centrico, randomizzato, in doppio cieco e placebo controllato che valuta sicurezza e tollerabilità di QRL-201 nella SLA. QRL-201 è un oligonucleotide antisenso che mira a ripristinare il gene Stathmin-2 (STMN2), i cui livelli sono ridotti nella SLA. La progettazione dello studio clinico è stata descritta nel poster CLT-08.
DNL343
DNL343 è un agonista del eIF2B coinvolto nella risposta allo stress che ha mostrato effetti protettivi in studi di laboratorio preclinici. In uno studio clinico di fase 1, descritto nel poster CLT-09, DNL343 è risultato sicuro e ben tollerato.
EPIC-ALS
EPIC-ALS è uno studio clinico multicentrico, a Gruppo singolo in fase 2 che include soltanto 10 pazienti con diagnosi definitiva di SLA. Lo studio, descritto nel poster CLT-10, valuta gli effetti di EPI-589 che sembra ridurre lo stress ossidativo.
IPL344
IPL344 è un attivatore di Akt, una proteina coinvolta in vie di trasduzione del segnale che promuovono la sopravvivenza cellulare. Attualmente, IPL344, come descritto nel poster CLT-12, è in sperimentazione in uno studio clinico in fase 1/2a per valutarne efficacia e sicurezza.
CLT-12: riferisce i risultati di sicurezza e tollerabilità dello studio di fase 1/2a su IPL344. L’IPL344 ha dimostrato di ridurre l’infiammazione e la morte cellulare, oltre a comportarsi in modo protettivo negli studi preclinici.
PARADIGM
PrimeC è una terapia combinata del farmaco anti-infiammatorio ciprofloxacina e dell’antibiotico colecoxib che è risultata efficace in modelli preclinici di SLA. PARADIGM è uno studio in fase 2b randomizzato, in doppio cieco e placebo controllato che valuta sicurezza, tollerabilità ed efficacia di PrimeC in persone che vivono con la SLA. Risultati preclinici e studio clinico sono stati descritti nel poster CLT-15.
Salbutamol
Salbutamol è in sperimentazione clinica in uno studio pilota di fase 2 atto a determinare le capacità di camminare nelle persone affette da SLA. Questo studio clinico ha la particolarità di avere come bersaglio la giunzione neuromuscolare, la cui funzione viene completamente persa nei pazienti SLA riducendo del tutto la capacità di camminare. Lo studio è stato descritto nel poster CLT-29.
Segui lo stato attuale e particolari di questi studi e qualsiasi altro in corso visitando l’elenco di sperimentazioni SLA-LIVE sul sito di conSLAncio.
<<>>
BIO-12 Caratterizzazione del ruolo dell’asse intestino-cervello nel fisiopatologia e progressione della malattia del motoneurone
Q. Khwaja1,2, I. Khwaja1, A. Ahmed1, L. Zampedri2, V. Lombardi2, S. Rowe3, A. Malaspina2
1Blizard Institute, Queen Mary University of London, Londra, Regno Unito;
2Institute of Neurology, University College London, Londra, Regno Unito;
3Basildon University Hospital, Basildon, Regno Unito
Background: La mancanza di predittori affidabili della progressione della malattia e di biomarcatori che misurino accuratamente la risposta al trattamento è una delle principali carenze nella gestione dei pazienti con MND e un importante ostacolo allo sviluppo di studi clinici convenienti. Le concentrazioni nel sangue e nel liquido cerebrospinale dei livelli di neurofilamenti a catena leggera (NfL) sono state studiate come biomarcatori prognostici in diversi studi, tuttavia nessun singolo marcatore riesce a dare un quadro completo della progressione della malattia in MND o che sia stato standardizzato e convalidato per l’uso in clinica.
Rappresentando il cross-talk bidirezionale tra il sistema nervoso enterico e il cervello, l’asse intestino-cervello (GBA) è recentemente emerso come potenziale implicato nella progressione della neurodegenerazione e un target per interventi terapeutici al fine di prevenire o rallentare la progressione della MND. Disregolazione di GBA cross-talk e comorbidità gastrointestinali significative sono state osservate in pazienti con malattie neurodegenerative, con una degenerazione che si verifica nell’intestino prima che nel sistema nervoso centrale. Pertanto postuliamo che la composizione del microbioma intestinale in pazienti con MND può promuovere o potenziare la risposta del sistema immunitario/infiammatorio che potrebbe non solo alterare la motilità intestinale ma anche svolgere un ruolo nella patogenesi della MND. La stitichezza è comunemente osservata nei pazienti con SLA e ritardi nello svuotamento gastrico e tempi di transito del colon sono stati segnalati anche in assenza di sintomi motori alterati, suggerendo una disfunzione autonomica.
Obiettivi: Lo scopo di questo studio osservazionale pilota è quello di indagare il ruolo del GBA nella progressione della MND attraverso la somministrazione di questionari che esaminano la prevalenza dei sintomi gastrointestinali nei pazienti con MND.
Metodi: Questionari per acquisire la progressione della malattia MND (tasso di progressione della malattia basato sulla SLA Functional Rating Scale-Revised [ALSFRS-R]) e sintomi gastrointestinali (sistema di punteggio della costipazione di Cleveland, Scala di valutazione dei sintomi gastrointestinali, deglutizione Questionario sui disturbi, scala Wexner per l’incontinenza, e indice di qualità della vita gastrointestinale) sono stati somministrati a 30 pazienti con MND in almeno 2 occasioni a intervalli di 6 mesi. Indagini demografiche e questionari sulla frequenza alimentare sono stati somministrati per comprenderne il pattern alimentare. ALSFRS-R cambia, insieme a fattori come l’età, sesso, modelli dietetici, insorgenza bulbare o degli arti e cambiamneti del BMI saranno correlati con la prevalenza dei sintomi gastrointestinali e livelli di NfL utilizzando un’analisi lineare multivariata con modello di regressione.
Risultato atteso: Si ipotizza che le interazioni intestino-cervello attraverso l’influenza di costituenti del microbioma intestinale sul sistema enterico, autonomo, neuroendocrino e pathway neuroimmuni, svolgono un ruolo chiave nella fisiopatologia della MND e influenzano il tasso di progressione. In questo studio pilota, esamineremo le prove dell’associazione tra modelli dietetici e sintomi gastrointestinali con lo stadio e il tasso di progressione di MND, stabilendo così il terreno per ulteriori studi che esaminano la fisiopatologia del il GBA in MND.
<<>>
IVT-04 Caratterizzazione del pathway PPIA/EMMPRIN in modelli di sclerosi laterale amiotrofica familiare
Gloria N. Edozie1, Shanna Pigeyre1, Ariane Gosselin1, Maria Del Carmen Pelaez Brenes1, Chantelle Sephton1, Silvia Pozzi1
1CERVO Brain Research Centre, Université Laval, Québec, QC, Canada.
Background: La neuroinfiammazione svolge un ruolo significativo nell’insorgenza e nella progressione della sclerosi laterale amiotrofica (SLA), che attualmente non ha un trattamento efficace. La proteina peptidilprolil cis-/trans-isomerasi A (PPIA), nota anche come ciclofilina A, è una proteina chaperone con attività di ripiegamento. Nei modelli di SOD1 G93A della SLA, PPIA è nota per il suo ruolo favorevole a livello intracellulare e per i suoi effetti svantaggiosi a livello extracellulare. L’interazione di PPIA con il suo recettore EMMPRIN (induttore di metalloproteasi della matrice) stimola infatti le vie infiammatorie guidate dal fattore nucleare kappa B e il rilascio di MMP9. La rilevanza di questa via nei casi di SLA diversi dalla SOD1 G93A è ancora sconosciuta.
Obiettivo: Valutare se la secrezione di PPIA e l’espressione di EMMPRIN siano simili nei diversi modelli murini di SLA familiare.
Metodi: I topi SOD1 G93A, TDP-43 A315T e Meox-Cre-FUS R5215G sono stati mantenuti presso il CERVO Brain Research Centre e sacrificati in diversi stadi della malattia insieme a topi non transgenici (NTg) appaiati per età e sesso. Il midollo spinale lombare (SCL) e il fluido cerebrospinale (CSF) sono stati raccolti, processati e analizzati mediante immunoblotting per EMMPRIN e PPIA. L’espressione di EMMPRIN è stata valutata anche mediante immunofluorescenza nel midollo spinale dei topi.
Risultati: Abbiamo riscontrato un aumento del rilascio di PPIA nel liquido cerebrospinale e dell’espressione di EMMPRIN nel midollo spinale di topi SOD1 G93A, confermando i dati precedentemente pubblicati. Nei topi TDP-43 A315T, abbiamo osservato un aumento significativo del rilascio di PPIA all’esordio della malattia, correlato a un aumento dell’espressione di EMMPRIN nel midollo spinale lombare di questi topi. La PPIA nel CSF di topi Meox-Cre-FUS R521G è ridotta rispetto ai topi NTg. Analizzando l’espressione di EMMPRIN nel midollo spinale dei topi SOD1 G93A e TDP-43 A315T, abbiamo riscontrato un’espressione specifica del recettore nei motoneuroni e negli astrociti.
Discussione: L’interazione tra PPIA extracellulare ed EMPPRIN è un interessante bersaglio terapeutico per la SLA. I nostri risultati dimostrano che questa via può essere bersagliata nelle forme familiari di SLA dipendenti da mutazioni dei geni SOD1 e TDP-43, mentre la sua rilevanza potrebbe essere minore nelle forme di SLA mediate da mutazioni sul gene FUS. Inoltre, nel midollo spinale di topi mutanti per SOD1 e TDP-43, abbiamo osservato che i motoneuroni e gli astrociti esprimono EMMPRIN, evidenziando la sensibilità di questi tipi di cellule alla via PPIA/EMMPRIN.
<<>>
IVT-13 Caratterizzazione del pathway PPIA/EMMPRIN nel sistema nervoso periferico di modelli SOD1G93A di sclerosi laterale amiotrofica
Marion Boyer1, Frédérique Crépeau1, Silvia Pozzi1
1CERVO Brain Research Centre, Université Laval, Québec, QC, Canada.
Background: La proteina peptidil-prolil isomerasi A (PPIA) è stata identificata come biomarcatore traslazionale per la sclerosi laterale amiotrofica (SLA). PPIA è una proteina chaperone con attività cis-trans isomerasica coinvolta nel ripiegamento e ell’assemblaggio delle proteine. Nel sistema nervoso centrale dei topi SOD1G93A questa proteina è altamente espressa dai motoneuroni e dalle cellule gliali. Una volta rilasciata, PPIA extracellulare (ePPIA) si lega al suo recettore EMMPRIN inducendo eventi proinfiammatori e il rilascio di metalloproteasi della matrice (MMP). È noto che la MMP-9 sia implicata nella morte dei motoneuroni e che MMP-7 abbia un ruolo nella migrazione delle cellule di Schwann e nella mielinizzazione degli assoni. Il ruolo di PPIA e della via PPIA/EMMPRIN nel sistema nervoso periferico (PNS) della SLA rimane ancora sconosciuto.
Obiettivi: Questo studio intende decifrare la via PPIA/EMMPRIN nel PNS di un modello animale di SLA ben caratterizzato, il topo SOD1G93A. Abbiamo ipotizzato che PPIA sia implicata nelle vie di comunicazione tra le cellule di Schwann, i motoneuroni e le cellule muscolari e che PPIA possa contribuire alla degenerazione dei motoneuroni e all’attivazione delle cellule gliali nel sistema nervoso periferico in condizioni di SLA.
Metodi: Abbiamo raccolto nervi sciatici (SN) e muscoli tibiali anteriori (TA) di topi non transgenici (Ntg) e da topi SOD1G93A a diversi stadi della malattia. Abbiamo eseguito esperimenti di immunofluorescenza per scoprire la localizzazione di PPIA e EMMPRIN, ed esperimenti di western blot per studiare il livello e l’aggregazione di PPIA nei tessuti del sistema nervoso periferico.
Risultati: Le analisi di Western blot hanno mostrato che l’espressione di PPIA è alterata nei tessuti del sistema nervoso periferico dei topi SOD1G93A con la progressione della malattia, sia come proteina solubile che insolubile. Gli esperimenti di immunofluorescenza hanno rivelato la presenza di PPIA ed EMMPRIN in diverse popolazioni cellulari del PNS.
Discussione: Sebbene preliminare, questo studio mostra alterazioni dei livelli di PPIA nel sistema nervoso periferico dei topi SOD1G93A. Data la rilevanza di PPIA nel sistema nervoso centrale dei topi SOD1G93A, sia come proteina intracellulare che extracellulare, i nostri risultati evidenziano una possibile implicazione di PPIA e della via PPIA/EMMPRIN anche nel sistema nervoso periferico.
<<>>
DSP-11 La caratterizzazione trascrittomica dei fenotipi di SLA ha evidenziato un pattern di espressione paziente-specifico
M. Garofalo1, E. Scarian1,2, F. Dragoni1,3, R. Di Gerlando1,3, L. Grieco3, M. Busacca3, G. Fiamingo3, L. Diamanti1, J. Garau1, M. Bordoni1, O. Pansarasa1, S. Gagliardi1
1IRCCS Mondino Foundation, Pavia, Italy
2University of Pavia, Dep. of Brain and Behavioral Sciences, Pavia, Italy
3University of Pavia, Dep. of Biology and Biotechnology “L. Spallanzani”, Pavia, Italy
Riassunto: Esigenza critica nella ricerca sulla SLA è l’identificazione sia di una strategia adeguata per stratificare i pazienti sia di biomarcatori affidabili. L’alterazione dell’espressione genica potrebbe rappresentare una nuova opportunità per identificare tratti paziente-specifici. Infatti, la classificazione basata su fenotipi clinici potrebbe essere associata a diversi modelli di espressione genica, rappresentando una nuova opportunità per caratterizzare sottotipi di SLA con caratteristiche cliniche e biologiche omogenee.
Il nostro obiettivo è identificare il profilo trascrittomico di diversi fenotipi SLA e utilizzare queste informazioni per la valutazione dei biomarcatori e lo sviluppo di una eventuale terapia personale.
Abbiamo caratterizzato 48 pazienti affetti da SLA sporadica: “Classic” (n=12), “Bulbar” (n=10), “Flail Arm” (n=7), “Flail Leg” (n=10) e “piramidale” (n=9). Quindi, è stato effettuato il sequenziamento dell’RNA estratto dai PBMC isolati dai pazienti e dai controlli sani (n=19).
Abbiamo osservato un pattern di espressione genica distintivo tra pazienti e controlli, in particolare per il gruppo “Flail Leg”. Inoltre, il gruppo “Bulbar” era quello che mostrava la maggiore quantità di geni alterati, coerentemente con le caratteristiche peculiari di questo fenotipo. È interessante notare che abbiamo trovato un solo gene comunemente deregolato in tutti i gruppi, mentre il resto dei geni erano specifici del fenotipo. Inoltre, abbiamo osservato una forte deregolazione dei geni coinvolti nei pathway infiammatori. Pertanto, al fine di monitorare la progressione della malattia e definire fattori diagnostici e prognostici, miriamo a includere un’analisi di follow-up a un anno.
In conclusione, l’identificazione dei meccanismi patogenetici fenotipo-specifici sarà cruciale per l’identificazione di marcatori di progressione.
<<>>
CMS-33 La registrazione dei movimenti oculari:
una nuova fonte di informazioni cliniche nella SLA
Cozza F1,2,3, Lizio A1, Garassino G4, Greco L1,2, Casiraghi J1, Lunetta C1,5, Sansone V1,6, Cerri F1,2
1Neuromuscular Omnicentre (NEMO), Fondazione Serena Onlus, Milan, Italy
2Nemo Lab, Milan, Italy
3COMiB Research Centre in Optics and Optometry, University of Milano-Bicocca, Milan, Italy
4Department of Materials Science, University of Milano-Bicocca, Milan, Italy
5Neurorehabilitation Department, Istituti Clinici Scientifici Maugeri, IRCCS, Milan, Italy
6Neurorehabilitation Unit, Department of Biomedical Sciences of Health, University of Milan, Milan, Italy
Introduzione e obiettivi: Negli stadi più avanzati della Sclerosi Laterale Amiotrofica, avere una visione nitida e un’accurata motilità oculare è un aspetto cruciale per i pazienti che abitualmente comunicano tramite sistemi a puntamento oculare.
Il sistema visivo è generalmente preservato dalla patologia, tuttavia alcuni recenti studi riportano la presenza di deficit oculari in diversi stadi della malattia, soprattutto a carico della funzione oculomotoria.
Lo scopo di questo studio è quello di valutare i movimenti oculari e di indagarne la relazione con la compromissione clinica, utilizzando un eye tracker ad alta frequenza.
Metodi: Sono stati arruolati pazienti con SLA definita, probabile o probabile supportata da esami dilaboratorio sulla base dei criteri El Escorial.
Per ogni paziente è stata svolta una valutazione clinica e optometrica ed è stata somministrata la
sequenza di tasks tramite eye tracker.
Le variabili cliniche raccolte sono la scala Amyotrophic Lateral Sclerosis Functional Rating Scale–revised (ALSFRS-r), il sito di esordio, l’indice di progressione della patologia, l’uso della ventilazione invasiva e non invasiva, e la gastrostomia alla valutazione.
I dati optometrici includono il test della motilità oculare (EOM), il Northeastern State University
College of Optometry (NSUCO) che valuta le saccadi e gli inseguimenti, l’errore refrattivo e la
valutazione della visione binoculare tramite cover test e test della stereopsi.
I movimenti oculari sono stati registrati tramite uno screen-based eye tracker alla frequenza di 600 Hz: sono state indagate la fissazione, le saccadi, le anti-saccadi orizzontali e verticali, e le saccadi delayed. Nello specifico, nel task di fissazione e delle saccadi è stato rilevato il tempo di fissazione nei target definiti dalle aree di interesse. Per quanto riguarda il task delle anti-saccadi e saccadi delayed, l’error rate (%) è il principale outcome indagato.
Risultati: Sono stati arruolati 40 pazienti affetti da SLA con età mediana di 59.32 anni [52.07 – 67.14] e rapporto M/F di 2.08 (M/F: 27/13).
I pazienti più compromessi a livello funzionale, secondo il punteggio totale e quello dei singoli
domini (bulbare, motorio e respiratorio) della scala ALSFRS-r, hanno commesso un numero di errori significativamente maggiore nel test anti-saccadi orizzontali e verticali, e nelle saccadi delayed.
Inoltre, una maggior compromissione funzionale espressa dalla scala ALSFRS-r risulta essere
significativamente associata a movimenti oculari meno accurati nel task della fissazione; la stessa tendenza emerge nel task delle saccadi, sebbene non sia statisticamente significativa.
Infine, dal confronto fra i dati eye tracker e le variabili optometriche emerge come i pazienti con
una peggiore performance nei test NSUCO e con movimenti oculari meno accurati nell’EOM test abbiano un error rate significativamente più alto nei test di anti-saccadi e saccadi delayed.
Conclusioni: I nostri risultati mostrano che la compromissione funzionale espressa dalla scala ALSFRS-r è associata ad aspetti specifici della funzione oculomotoria nei pazienti SLA, in linea con la letteratura. Come e perché il deficit oculare sia correlato alla progressione della patologia è un aspetto che merita ulteriori approfondimenti, così come l’influenza di eventuali deficit cognitivi sui test proposti all’eye tracker. Attualmente è in corso uno studio longitudinale sui pazienti SLA per rilevare possibili variazioni nei risultati dell’eye tracker col progredire della patologia.
<<>>
COG-12 Versione Italiana della Hospital Anxiety and Depression Scale per le Malattie del Motoneurone: indagare il disturbo dell’umore in un ampia corte di pazienti con MND
Monica Consonni1, Veronica Faltracco1, Debora Pain2 , Alessandra Telesca1,3, Eleonora Dalla Bella1, Enrica Bersano1,4, Elisabetta Soldini1, Gabriele Mora2, Giuseppe Lauria1,4
1Fondazione IRCCS Istituto Neurologico Carlo Besta
2Istituti Clinici Scientifici Maugeri, IRCCS
3Department of Medicine and Surgery, University of Milano-Bicocca, Milan
4Department of Medical Biotechnology and Translational Medicine, University of Milan
Riassunto: La Hospital Anxiety and Depression Scale per le Malattie del Motoneurone (HADS-MND) è una scala per la valutazione dei sintomi di ansia e depressione nei pazienti con MND. Ad oggi non esiste una validazione della versione italiana (itHADS-MND) che possa essere usata come screening per misurare l’assetto timico dei pazienti e per studiare la correlazione con le loro disabilità motoria e il deterioramento cognitivo-comportamentale. A tal fine 275 pazienti con MND hanno compilato la it-HADS-MND ed eseguito una valutazione neuropsicologica mirata all’identificazione del profilo cognitivo-comportamentale e della sintomatologia ansiosa (State Anxiety Inventory> 50) e depressiva (Beck Depression Inventory > 20). La struttura fattoriale della it-HADS-MND è caratterizzata da due componenti distinte che sottendono le sotto-scale di ansia (6 items; α=0,73) e depressione (6 items; α=0,82). Le due sotto-scale non hanno però sufficiente specificità, come emerso dalle curve Receiver Operating Characteristics, nell’identificare ansia e depressione. Diversamente, il punteggio globale della itHADS-MND, i cui items hanno un’elevata consistenza interna (α=0.85), ha sufficiente potere discriminante (AUC=0,83; IC 95%=0.78–0.87; cutoff=10: sensibilità 73%; specificità 77%) per riscontrare globalmente una sintomatologia ansioso-depressiva. Sintomatologia che è presente in 89 pazienti. Le analisi di correlazione hanno confermato la validità della scala come misura dell’assetto timico poiché associata a misure del funzionamento affettivo-emotivo e della disabilità motoria (p<0. 005). La debolezza delle correlazioni con il funzionamento cognitivo-comportamentale suggerisce invece che la itHADS-MND è parzialmente indipendente dai disturbi dello spettro frontotemporale.
<<>>
Il simposio si è concluso con l’assegnazione di due ulteriori premi:
 Il Premio Healey Center International Prize for Innovation in ALS 2022. Questo premio di 50.000 dollari è stato conferito al team che ha contributo all’approvazione del farmaco AMX0035 negli Stati Uniti e Canada: Josh Cohen, CEO e co-fondatore dell’Amylyx Pharmaceuticals, Justin Klee, Presidente e co-fondatore dell’Amylyx Pharmaceuticals, Kent Leslie, Direttore Scientifico dell’Amylyx Pharmaceuticals, Dott.ssa Sabrina Paganoni, M.D., PhD, Capo Investigatore dello studio CENTAUR, Co-direttore del Massachusetts General Hospital Neurological Clinical Research Institute (MGH NCRI), Dott. James Berry, M.D., MPH, Co-direttore del Consorzio NEALS e direttore del MGH NCRI, Dott.ssa Jinsy Andrews, M.D., MSc., Co-direttore del Consorzio NEALS e il Dott. Jeremy Shefner, M.D., PhD, Senior Vice Presidente, Barrow Neurological Institute (BNI). Il Premio Healey Center International Prize for Innovation in ALS viene assegnato ogni anno per riconoscere il lavoro “che porta a scoperte e progressi eccezionali nello sviluppo di terapia per la SLA.”
Il Premio Healey Center International Prize for Innovation in ALS 2022. Questo premio di 50.000 dollari è stato conferito al team che ha contributo all’approvazione del farmaco AMX0035 negli Stati Uniti e Canada: Josh Cohen, CEO e co-fondatore dell’Amylyx Pharmaceuticals, Justin Klee, Presidente e co-fondatore dell’Amylyx Pharmaceuticals, Kent Leslie, Direttore Scientifico dell’Amylyx Pharmaceuticals, Dott.ssa Sabrina Paganoni, M.D., PhD, Capo Investigatore dello studio CENTAUR, Co-direttore del Massachusetts General Hospital Neurological Clinical Research Institute (MGH NCRI), Dott. James Berry, M.D., MPH, Co-direttore del Consorzio NEALS e direttore del MGH NCRI, Dott.ssa Jinsy Andrews, M.D., MSc., Co-direttore del Consorzio NEALS e il Dott. Jeremy Shefner, M.D., PhD, Senior Vice Presidente, Barrow Neurological Institute (BNI). Il Premio Healey Center International Prize for Innovation in ALS viene assegnato ogni anno per riconoscere il lavoro “che porta a scoperte e progressi eccezionali nello sviluppo di terapia per la SLA.”
L’ultimo premio:
 Il Drs. Ayeez and Shelena Lalji & Family ALS Endowed Award for Innovative Healing Award 2022 è stato conferito a due gruppi scientifici:
Il Drs. Ayeez and Shelena Lalji & Family ALS Endowed Award for Innovative Healing Award 2022 è stato conferito a due gruppi scientifici:
 1) Al prof. Jeffrey Rothstein, Direttore, Robert Packard Center for ALS Research, Professore di neurologia presso la John Hopkins University, USA e al prof. Clive N. Svendsen, Professore di scienze biomediche presso l’ospedale Cedars-Sinai Medical Center, USA, entrambi del gruppo scientifico Answer ALS per lo sviluppo di un trattamento sperimentale che combina la terapia genica con le cellule staminali. Lo studio intitolato, “Transplantation of human neural progenitor cells secreting GDNF into the spinal cord of patients with ALS: a phase 1/2a trial” è stato pubblicato sulla rivista Nature Medicine nel 2022.
1) Al prof. Jeffrey Rothstein, Direttore, Robert Packard Center for ALS Research, Professore di neurologia presso la John Hopkins University, USA e al prof. Clive N. Svendsen, Professore di scienze biomediche presso l’ospedale Cedars-Sinai Medical Center, USA, entrambi del gruppo scientifico Answer ALS per lo sviluppo di un trattamento sperimentale che combina la terapia genica con le cellule staminali. Lo studio intitolato, “Transplantation of human neural progenitor cells secreting GDNF into the spinal cord of patients with ALS: a phase 1/2a trial” è stato pubblicato sulla rivista Nature Medicine nel 2022.
 2) Il team dello studio Stathmin-2: Dott.ssa Clotilde Lagier-Tourenne, Harvard Medical School, Dott. Don Cleveland della University of California San Diego (UCSD), Dott. Jone Lopez-Erauskin dell’UCSD, Dott. Zevik Melamed dell’UCSD e il Dott. Kevin Eggan di BioMarin Pharmaceutical per il loro lavoro sulla regenerazione degli assoni. I due gruppi hanno vinto un finanziamento di 40.000 dollari. Il premio, assegnato per il secondo anno durante il simposio, è stato conferito dalla Dott.ssa Merit Cudkowicz, M.D., MSc., Direttore del Sean M. Healey & AMG Center for ALS presso il Massachusetts General Hospital, e da i Dott.ri Ayeez e Shelena Lalji.
2) Il team dello studio Stathmin-2: Dott.ssa Clotilde Lagier-Tourenne, Harvard Medical School, Dott. Don Cleveland della University of California San Diego (UCSD), Dott. Jone Lopez-Erauskin dell’UCSD, Dott. Zevik Melamed dell’UCSD e il Dott. Kevin Eggan di BioMarin Pharmaceutical per il loro lavoro sulla regenerazione degli assoni. I due gruppi hanno vinto un finanziamento di 40.000 dollari. Il premio, assegnato per il secondo anno durante il simposio, è stato conferito dalla Dott.ssa Merit Cudkowicz, M.D., MSc., Direttore del Sean M. Healey & AMG Center for ALS presso il Massachusetts General Hospital, e da i Dott.ri Ayeez e Shelena Lalji.
<<>>
conSLAncio ringrazia, a nome di tutti gli associati e di tutte le persone affette da SLA che seguono i nostri canali.
Il prossimo 34° Simposio Internazionale sulla SLA/MND avrà luogo presso Basilea, Svizzera dal 6-8 Dicembre 2023.
.
Per ulteriori informazioni su conSLAncio, visita: www.conslancio.it
Contatto:
L’ufficio stampa di conSLAncio, scrivere: info@conslancio.it
Risorse correlate:
| File | Descrizione | Dim. |
|---|---|---|
 conSLAncio_ALSMNDS-Poster2022-1-pdf
conSLAncio_ALSMNDS-Poster2022-1-pdf
|
1 MB |